Copyright: J. E. Wampler, 1996
This section covers:
Interactions in proteins.
The hydrophobic effect and protein structure.
Characteristics of the folded protein.
Folding motifs.
Covalent bonds have been discussed in Section I of this tutorial under amide bonds and crosslinks.
In vacuo, two point charges interact as described by Coulomb's law:
For a protein in solution, the interactions of charges are more complex than for two charged points in a vacuum because the dielectric constant varies from water (E~80) to the protein interior (E ~ 4). Here the interaction energy is described by the Poisson-Boltzmann equation.
If two oppositely charged atoms are free to move under the force of their attraction, they are drawn into "contact" as defined by the sharply rising repulsion that occurs as their electron clouds start to overlap. As a first approximation, this closest approach distance can be viewed as the contact of hard spheres with radii determined by the atomic number and electronic configuration of the atoms, i. e. the atomic or ionic radii. These radii are often referred to as van der Waal's radii or non-bonded radii.
As the non-bonded interaction between atoms and groups involves less than full formal charge and involves polarization contributions, the distance dependence falls of more quickly than the 1/r dependence of Coulomb's law. In these more complicated cases, where the charges can not be represented by single point locations, the interactions are also less isotropic, falling off not just as a function of distance, but also as a function of orientation:
Distance and Angle Dependence of Non-bonded Interactions.
With fixed magnitude charges:
POINT CHARGE with POINT CHARGE 1/r
POINT CHARGE with DIPOLE Cos (angle) x 1/r2
DIPOLE with DIPOLE F(angle)* x 1/r3
POINT CHARGE with QUADRAPOLE ~1/r3
DIPOLE with QUADRAPOLE ~1/r4
QUADRAPOLE with QUADRAPOLE ~1/r5
With polarizable charge centers:
POINT CHARGE w/ POLARIZABLE DIPOLE ~ 1/r4
INDUCED DIPOLE-DIPOLE ~ 1/r6
INDUCED DIPOLE-OCTUPOLE ~ 1/r8
INDUCED QUADRAPOLE-QUADRAPOLE ~ 1/r10
* where F(angle) is a function of the cosines and sines of the angles between the dipole moments and the separation vector.
Hydrogen-bonding:The database of know structure can also give us information about the potential interactions within a protein that help maintain its conformation. J. Singh & J. M. Thornton published an " Atlas of Protein Side-Chain Interactions" (Vols. I & II, IRL Press, Oxford, 1992) which is now available on-line in an updated version.
Hydrogen bound to Oxygen, Nitrogen and Sulfur has a smaller van der Waal's radius and more partial charge than hydrogen on carbon making the interaction between a charge or dipole with such "donar groups" stronger and more orientation dependent.Aromatic-Aromatic Interactions:
The aromatic amino acid sidechains of phenylalanine, tyrosine and tryptophane prefer interactions with interplane angles of around 90o. The interaction has a quadrupole-quadrupole character (distance dependence ~1/r5) with dependence on the interplane angle.Aromatic-Polar/Charged-Group Interactions:
The charge separation of aromatics with ring hydrogens having partial positive charges and the pi electron system being partially negative, leads to interactions with O, S and N groups in proteins which are relatively strong (distance dependence ~1/r3 to 1/r4) and orientation dependent.
A similar " Atlas of Side-Chain and Main-Chain Hydrogen Bonding" is also available, published by I. McDonald and J. M. Thornton.
Natural variation from species-to-species tends to favor changes in surface (and therefore polar) groups.
Structure is determined globally and redundantly. Upto 30% of the amino acids in some proteins have been changed to alanine with little change in the folded structure.
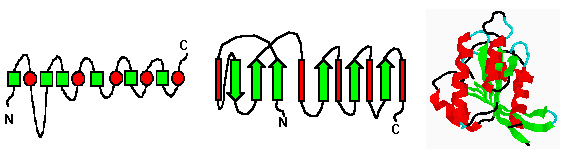
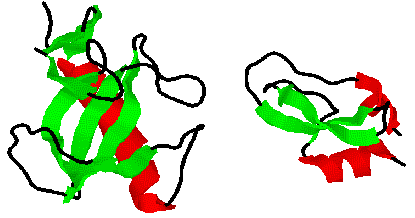
At the simplest level, proteins can be classified by their content of
secondary structure. Typically, the focus is on alpha-helicies and beta-
sheets. However, there are some proteins where turns and disulfide bonds
seem to be more important considerations.

Consider wheat germ agglutinin, where 16 turns (in cyan) and 16 disulfide
bonds (in brown) are the predominant structural contributions. While a
large part of the structure has no classified secondary structure (in black),
there are still four structurally homologous domains related by symmetry.
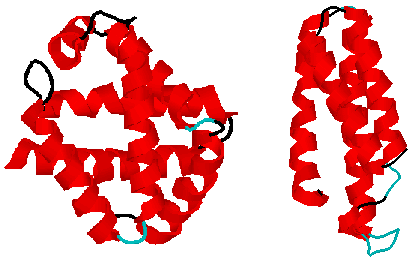
Some proteins are made up of mostly alpha helicies:
Some are mostly beta sheet: